Haplotype-Lso Tutorial¶
For this tutorial, first download the examples ZIP file and extract it to your computer.
Upload Files¶
Next, use the “upload files” button on the upper right to upload the files.
Navigate to where you extracted the files. Then select all files (e.g., by pressing Ctrl + a at the same time) and upload all files.
Select the files you wan to upload (Ctrl + a to select all files).
Wait for a moment and the haplotyping results will be displayed.
Note
When looking at the example data, you will notice that the file names all follow the same pattern:
The first element of the file name is the sample name, the second element is the name of the target region, separated by a dot. The tool will use this information to group your sequences later and (a) group type information by sample and (b) compute consensus by sample and region. Item (a) allows to determine the haplotype based on multipe regions per sample and (b) allows to use sequence from both forward and reverse primers.
Optionally, you can also add more information (e.g., primer-related) in a third group: <sample>.<region>.<primer>.fasta.
Result Summary Tab¶
Note that you can download the results information also as an Excel file using the “Download XLSX” links on the top.
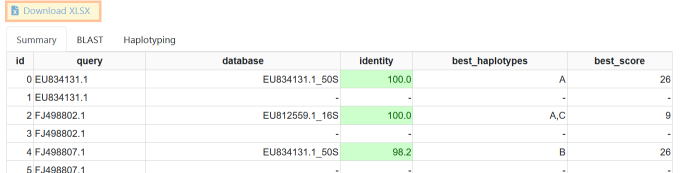
Result summary tab.
The Summary tab shows the following information:
- query
- the query file name
- database
- the ID of the database sequence with the best match.
The name will start with the GenBank identifier, followed by an underscore
_and then the name of the region (currently one of 16S, 16S-23S, or 50S). - identity
- the identity of the BLAST match with the reference in percent.
- best_haplotypes
- the best haplotypes based on the informative values on the reference. NB: if the sequencing error rate is high or the sequence is too short then not all informative positions will be covered. In this case, there can be ambiguity and more than one haplotype can be returned. For example, haplotypes A and C only differ in a single position on the 16S locus.
- best_score
- the score of the best match used for haplotype identification. Concordance with a variant in the haplotyping table contributes a score of “plus one”, discordance contributes a “minus one”. The sum is the overall score.
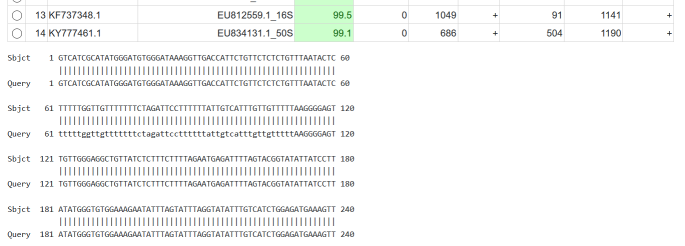
Result BLAST Tab¶
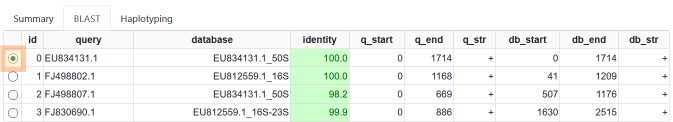
The BLAST result tab.
The BLAST tab provides the following information:
- query, database, identity
- see above
- q_start, q_end, q_str
- the start and end position of the match in the query and its strand
- db_start, db_end, db_start
- the start end end position of the match in the database and its strand
Further, you can select each match with the little round button on the right. The corresponding BLAST match will be displayed below the results table.
Result Haplotyping¶
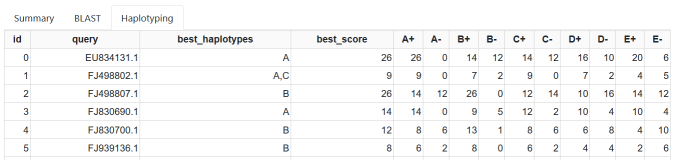
The haplotyping result tab.
The Haplotyping tab shows more details on the haplotyping results.
- query
- see above
- best_haplotypes
- the best haplotype(s) for the given query
- best_score
- the best score of the best haplotype(s)
- A+, A-, etc.
- for each haplotype known to Haplotype-LSO, the number of positive/concordant and negative/discordant position
Phylogenetic Analysis¶
The Dendrograms tab shows results of hierarchical clustering using the UPGMA algorithm for each region.
The input of the UPGMA algorithm is based on the pairwise BLAST identities (1.0 - identity).
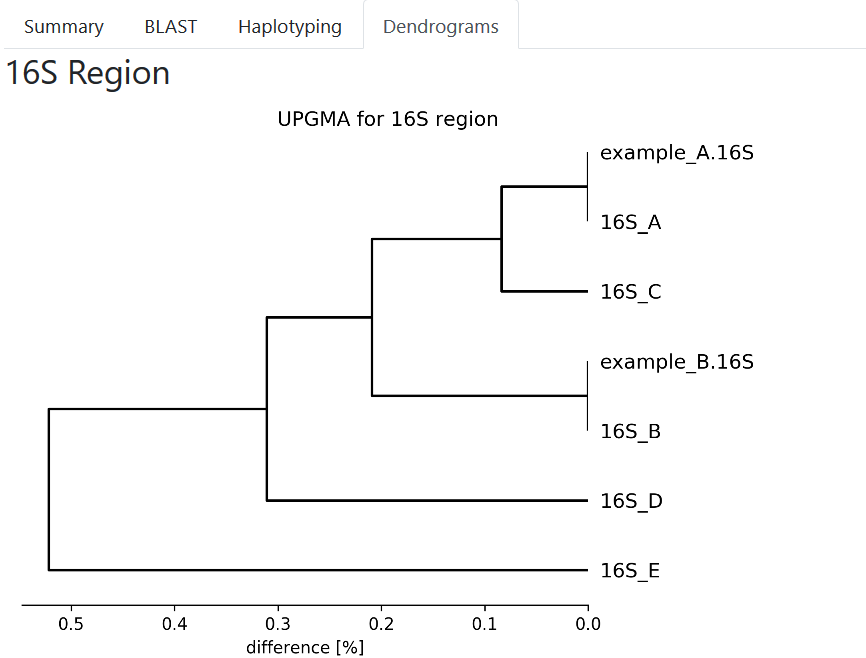
The dendrograms tab with the phylogenetics analysis.